

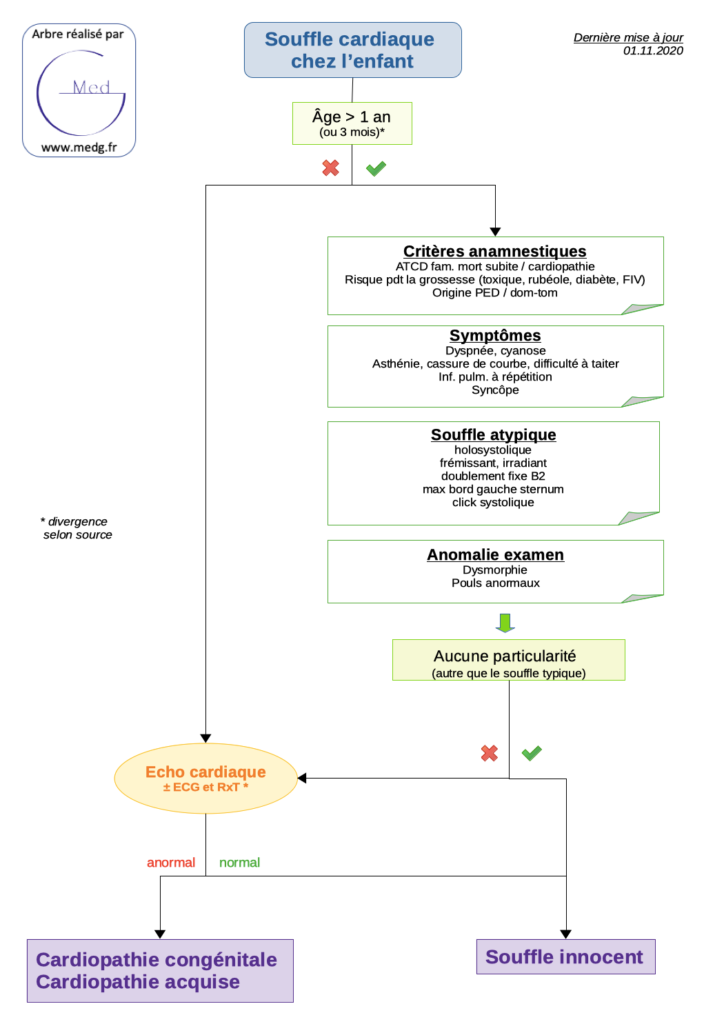
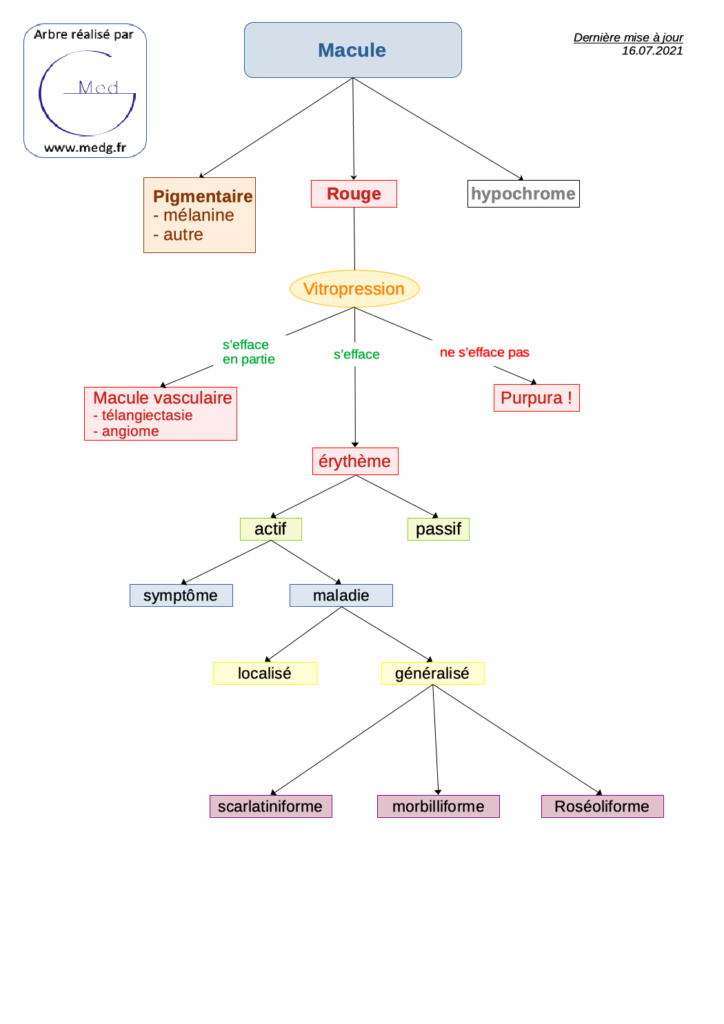
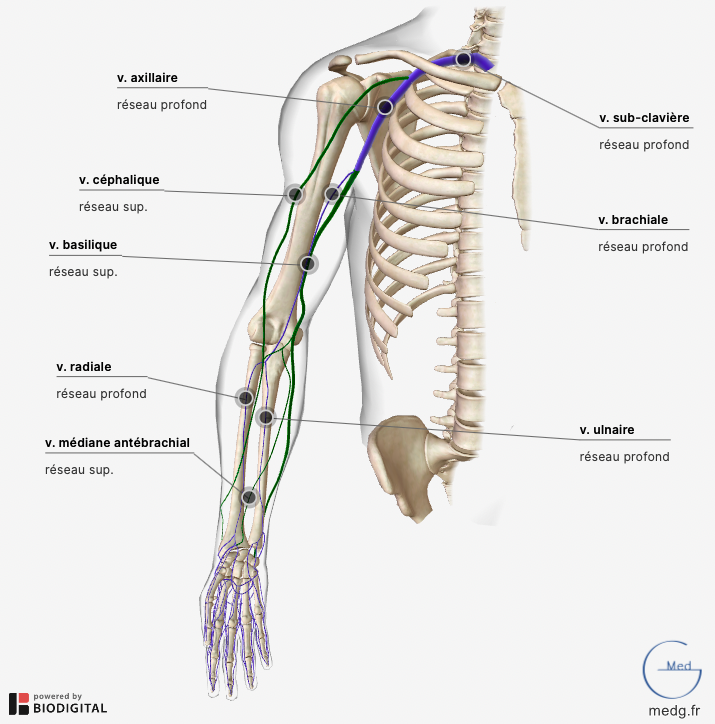
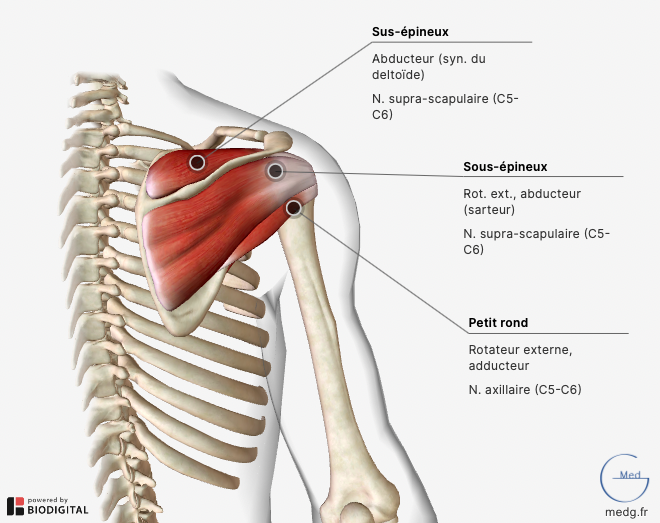
Actualités diverses
Juin 2022 : (Re)-découvrez le projet MedG, réécrit et détaillé.
Juin 2022 : Nouvelle mise à jour importante « sous le capot » avec amélioration de la navigation, correction de bugs et ajout de fonctionnalités :
– mode sombre pour les travailleurs du soir (en bas à droite)
– mode zen : fiche en plein écran sur ordi (dans le menu navigation)
– surlignage des dernières mises à jour : à venir sur les prochaines MaJ de fiche (bouton à coté de « dernière mise à jour)
– fonction recherche développée, avec possibilité de chercher des liens externes dans la bibliothèque
– favori : ajout possible de liens externes en favori, qui sont alors surlignés en rouge.
Février 2022 : Asso reconnue d’intérêt générale !
C’est officiel ! Après plusieurs mois d’attente et un premier refus, l’association MedG.fr est reconnue d’intérêt général !
Janvier 2022 : Statistique de 2021
=> 450.000 visites !
Mai 2021 : Faites un don avec HelloAsso !
Après plusieurs mois de pause, vous pouvez de nouveau faire un don à l’asso MedG.fr. Vos dons sont exclusivement utilisé pour le développement du site. Nous avons besoin de vous ! En savoir plus.
Avril 2021 : création de l’association MedG.fr 🙂
Nouvelle grande étape dans le développement du site : il est maintenant géré par l’association MedG.fr ! N’hésitez-pas à nous rejoindre !
….