
1) Généralités 2
Déf : le syndrome de Joubert (SJ) est une maladie génétique de la famille des ciliopathies. Sa définition est neuro-radiologique : association d’une hypotonie, d’un retard de développement et d’une malformation cérébelleuse / du tronc cérébral spécifique à l’origine du signe de la molaire. Le SJ est caractérisé par une grande variabilité intra- et inter-familiale.
Physiopathologie : tous les gènes impliqués codent pour une protéine localisée dans le cil cellulaire primaire et/ou le corpuscule basal, ou dans le centrosome qui donne naissance aux cils primaires. Les cils primaires jouent un rôle dans la sensibilité chimique et mécanique à certaines voies de signalisation permettant la différenciation, division et polarisation cellulaire, leur dysfonction est à l’origine des différents signes observés dans le SJ et les autres ciliopathies. Le lien entre l’atteinte d’un gène de ciliopathie et les phénotypes spécifiques associés n’est pas élucidé.
La voie de signalisation Sonic HedgeHog (SHH) est critique dans la différenciation dorso-ventrale du tube neural, et dans la prolifération celullaire du granule cérébelleux, elle a une importance particulière dans la malformation spécifique de la fosse postérieure observée dans le SJ.
Epidémiologie : prévalence d’environ 1 / 100.000
Etiologies : au moins 33 gènes identifiés avec transmission autosomique récessive (AHI1, CPLANE1, CC2D2A, CEP290, TMEM67…) et 1 lié à l’X (OFD1).
2) Diagnostic 2
| Clinique | Paraclinique |
|---|---|
| Hypotonie puis ataxie Retard de développement / Déficience intellectuelle |
Imagerie cérébrale : signe de la molaire |
A ) Clinique
-
Examen physique
NB : les signes et leur intensité sont très variables d’une famille à l’autre, et au sein d’une même famille.
Signes fréquents
– Hypotonie dans la petite enfance (critère diagnostique) puis développement d’une ataxie
– Retard de développement / déficience intellectuelle (critère diagnostique) *, apraxie verbale
– Troubles respiratoires : tachypnée et/ou apnée pouvant alterner, apnées du sommeil
– Troubles oculomoteurs : apraxie, difficultés de poursuite visuelle, nystagmus
* La déficience intellectuelle est d’intensité variable, elle peut être absente ou sévère. D’autres troubles neuro-développementaux ont été décrits (TSA, TDAH, troubles du comportement…)
Signes plus rares (<50%)
– Ophtalmo : rétinite pigmentaire, colobome, ptosis
– Néphro : néphronoptyse, dysplasie kystique
– Neuro : épilepsie
– Digestif : fibrose hépatique, maladie de Hirschprung
– Anomalies des membres et orthopédiques: polydactylie souvent post-axiale, scoliose
– ORL : hamartomes oraux, fente, macroglossie, surdité de perception, ou de transmission du fait d’infections répétées
– Troubles endocriniens : déficit central isolé ou panhypopituitarisme, micropénis, obésité
– Cardio : communication inter-ventriculaire / atriale, coactation de l’aorte, anomalies de la valve aortique
– Situs invertus
Eléments dysmorphiques
– Visage allongé avec rétrécissement bi-temporal
– Télécanthus, sourcils archés
– Oreilles bas implantées, ailes du nez hypoplasiques avec narines antéversées
– Bouche triangulaire, micrognatie, prognatie
-
Formes cliniques
Des sous-types cliniques ont été décrits selon les signes associés (non-développés ici). Les signes fréquents décrits plus haut et le signe de la molaire en imagerie sont observables quel que soit le sous-type.
Anciennement, certaines associations ont été décrites sous des noms de syndrome différents, et parfois regroupées sous le terme Joubert Syndrome Related Disorders. Ces entités sont désormais toutes reconnues comme des SJ
– Syndrome de Dekaban-Arima : rétinopathy, dysplasie kystique des reins
– Syndrome de Senior-Loken : rétinopathy, néphronoptise juvénile
– Syndrome COACH : hypoplasie vermienne Cérébelleuse, Oligophrénie, Ataxie, Colobome, Hepatic fibrosis
– Syndrome de Varadi-Papp = sd oro-facio-digital type VI incluant hypoplasie vermienne cérébelleuse, hamartomes de la langue, fente labiale, polydactylie centrale avec métacarpien en ‘Y’, atteinte rénale et cardiaque
On peut aussi décrire des spécificités par gène en cause :
| Gène | Rétinite | Colobome | Rein | Oculo-rénal | Foie | Oral | Polydactylie | Autres signes du JS pour ce gène | Autres phénotypes associés au gène |
|---|---|---|---|---|---|---|---|---|---|
| AHI1 | ++ | (+) | + | + | (+) | Polymicrogyrie | |||
| CPLANE1 | (+) | + | + | Sd oculo-facio-digital type VI Sd Meckel |
|||||
| CC2D2A | + | + | + | + | + | Encéphalocèle, hydrocéph, épilepsie | Sd Meckel | ||
| CEP290 | ++ | + | ++ | ++ | + | Encéphalocèle, cardiopathie, situs invertus | Amaurose congénitale de Leber Sd Meckel Sd Bardet-Biedl |
||
| CSPP1 | (+) | (+) | (+) | (+) | Surdité de perception, hypoplasie calleuse, encéphalocèle | Sd Meckel Sd Jeune |
|||
| INPP5E | + | + | + | (+) | + | Sd MORM | |||
| KIAA0586 | (+) | + | (+) | (+) | Expressivité variable ++ | Sd hydrolethalus Sd Jeune |
|||
| MKS1 | (+) | (+) | (+) | Sd Meckel Sd Bardet-Biedl |
|||||
| NPHP1 | + | ++ | + | Parfois signe de la molaire ‘mild’ | Sd Meckel Atteinte rétinienne Sd Cogan Sd Bardet-Biedl |
||||
| RPGRIP1L | (+) | (+) | ++ | + | (+) | (+) | Encéphalocèle | Sd Meckel | |
| TCTN2 | Pie bot | Sd Meckel | |||||||
| TMEM67 | + | + | ++ | (+) | Encéphalocèle | Sd Meckel | |||
| TMEM216 | (+) | (+) | ++ | + | (+) | + | + | Cardiopathie Encéphalocèle |
Sd Meckel |
B ) Paraclinique
Imagerie cérébrale (IRM / TDM)
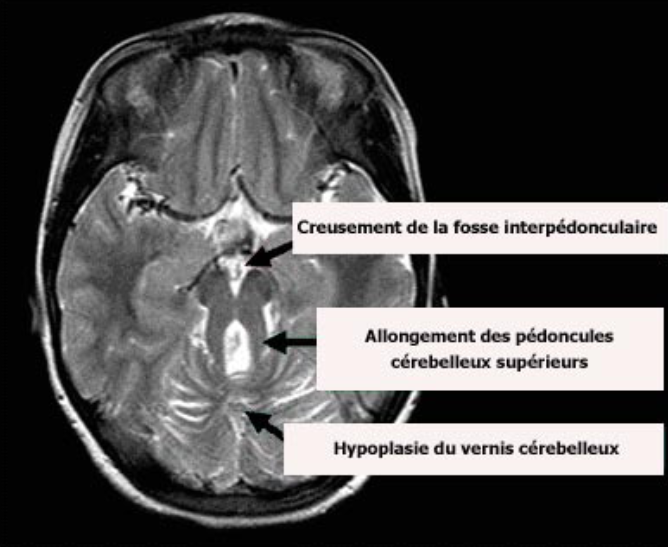
Sources :
– IRM tirée de www.genetests.org. Copyright, University of Washington, Seattle
– Légende issue de l’article orpha.net (http://www.orpha.net/data/patho/Pub/fr/Joubert-FRfrPub1022v01.pdf)
Signe de la molaire (critère diagnostique). Association de
– Fosse interpédonculaire profonde
– Pédoncules cérébelleux supérieurs épais et allongés
– Hypoplasie du vermis cérébelleux
Autres
– Agénésie / dysgénésie du corps calleux
– Cervelet : élargissement hémisphérique / hétérotopies
– Encéphalocèle occipitale / méningocèle
– Collection de LCS dans le 4e ventricule / hydrocéphalie (différentiel Dandy-Walker)
– Malformation / malrotation hippocampique
– Cortex cérébral: hétérotopies, dysplasie, pachygyrie, polymicrogyrie
Echo rénale (inconstant) :
– Dysplasie kystique dans les formes avancées = reins atrophiques et cicatriciels hyperéchogènes avec kystes de la jonction cortico-médullaire
– Parfois aspect compatible avec une polykystose autosomique récessive
Diagnostic moléculaire : par panel ou séquençage d’exome / génome pour le cas index. Recherche spécifique des mutations familiales connues dans le cadre d’un diagnostic prénatal ou d’une ségrégation familiale.
C ) Diagnostic différentiel
De nombreux différentiels sont des syndromes différents décrits pour des gènes du SJ (« allelic disorders »). Il s’agit pour la plupart de ciliopathies, et il est souvent difficile de déterminer s’il s’agit véritablement de syndromes différents ou s’ils font partie du spectre phénotypique du SJ.
Acrocallosal syndrome – hydrolethalus syndrome : macrocéphalie, DI, agénésie du corps calleux, anomalies de la fosse postérieure, hypertélorisme, polydactylie. Signe de la molaire parfois présent. Causes communes SJ : KIF7, KIAA0586.
Syndrome de Bardet-Biedl : rétinopathie, obésité, polydactylie, DI, hypogonadisme (♂) / malformations génitales (♀), atteinte rénale et parfois hépatique. Causes communes SJ : CEP290, MKS1, NPHP1.
Syndrome de Cogan : apraxie oculomotrice, hypoplasie vermienne cérébelleuse, parfois néphronoptise. Causes communes JS : NPHP1.
Syndrome de Jeune = dysplasie thoracique asphyxiante : thorax long et étroit, petite taille, polydactylie, dysplasie kystique rénale, autres anomalies osseuses ; souvent létal dans l’enfance. Causes communes JS : CSPP1, KIAA0586.
Amaurose congénitale de Leber : dystrophie rétinienne sévère avec nystagmus, kératocône, signe oculodigital de Franceschetti (enfant se frottant / pressant les yeux). Cause commune JS : CEP290.
Syndrome de Mainzer-Saldino : dystrophie rétinienne, atteinte rénale (typiquement néphronoptise), épiphyses phalangiennes en cône ± hypoplasie cérébelleuse, fibrose hépatique, dolichocéphalie. Causes communes JS : IFT140, IFT172.
Syndrome de Meckel : maladie rénale kystique, anomalies de la fosse postérieure, malformations hépatiques avec fibrose ± polydactylie, hypoplasie du vermis cérébelleux ; généralement létal en période pré- ou péri-natale. Causes communes JS : au moins 18 gènes dont CEP290, CPLANE1, TMEM67, TMEM216, CC2D2A, MKS1, TCTN2, CSPP1.
Syndrome MORM : retard Mental, Obésité, dystrophie Rétinienne, Micropénis. Cause commune JS : INPP5E.
Syndrome oro-facio-digital
– Type 1 : langue lobée ou avec hamartomes, fente, anomalies dentaires ; hypertélorisme, micrognatie ; brachy-, syn- ou polydactylie ; kystes cérébraux, dysgénésie du corps calleux ou du cervelet ± Dandy-Walker, maladie rénale kystique ; DI. Cause commune JS : OFD1.
– Type 4 : polysyndactylie, dysplasie tibiale, dysplasie tibiale, kystes rénaux, parfois anomalies cérébrales évocatrices de JS dans les formes sévères (létales). Cause commune JS : TCTN3.
– Type 6 (maintenant défini comme une forme de JS!) : polydactylie mésaxiale avec métacarpe en ‘Y’, hypoplasie du vermis cérébélleux, lobulations / hamartomes de la langue, ± atteinte rénale ou cardiaque. Le signe de la molaire fait partie des critères diagnostiques. Causes communes JS : CPLANE1, TMEM216, TMEM107, OFD1.
3) Evolution 2
A) Histoire naturelle
Les troubles respiratoires et visuels tendent à s’améliorer avec l’âge. Les apnées du sommeil sont plutôt centrales dans la petite enfance, et d’origine obstructive plus tard du fait de la macroglossie, l’hypotonie et l’obésité.
L’atteinte rénale, lorsqu’elle est existe, est présente dans la 1ère ou la 2e décennie. L’insuffisance rénale terminale survient en moyenne à l’âge de 13 ans.
L’ataxie n’est pas présente initialement mais apparaît fréquemment au cours du temps.
4) PEC 2
A ) Bilan
Bilan initial individuel
| Bilan initial |
|---|
| Clinique – Evaluation du développement psycho-moteur – Evaluation neurologique, ophtalmo, respiratoire, du sommeil – Evaluation orthophoniste |
| Paraclinique – Imagerie cérébrale : rechercher les malformations associées – Bilan ophtalmo : clinique PEV, électrorétinogramme – Echo abdo : recherche des anomalies hépatiques et rénales – Bilan sanguin et urinaire pour la recherche d’atteinte rénale : urée-créat sanguins, NFS, test de concentration sur urines matinales – Bilan hépatique sanguin – Polysomnographie si signe d’appel – Bilan endocrinien si signe d’appel – Bilan radiologique osseux si signe d’appel |
Bilan familial
– Ségrégation des variants pathogènes dans la famille
– Conseil génétique : transmission autosomique récessive +++ (sauf 1 forme liée à l’X), risque de récurrence de 25% à chaque grossesse pour un couple avec un enfant atteint. Diagnostic prénatal possible.
B ) Traitement
Il n’existe aucun traitement curatif ou spécifique du SJ. La prise en charge est symptomatique.
C) Suivi
Le bilan de suivi pourrait comprendre :
– Consultation annuelle en neurologie, neuropsy, ophtalmo
– Echo abdo annuelle
– Bilan hépatique et rénal (sang et urines) annuels



