
Neuro
Fiche réalisée selon le plan OD
Item ECNi 103
!! URGENCES !!
| clinique | etio |
|---|---|
| Etat de mal épileptique | Cause lésionnelle aigüe (dont AVC, méningite)L |
Définition 1A : ensemble de manifestations cliniques brutales, imprévisibles te transitoires qui résultent de l’hyperactivité (hyperexcitabilité et hypersynchronie) paroxystique d’un réseau de neurones corticaux ou cortico-sous-corticaux ; et de son éventuelle propagation.
Diagnostic positif : cf fiche MGS : Crise épileptique
Remarque : les convulsions du nourrisson et de l’enfant font l’objet d’une fiche dédiée
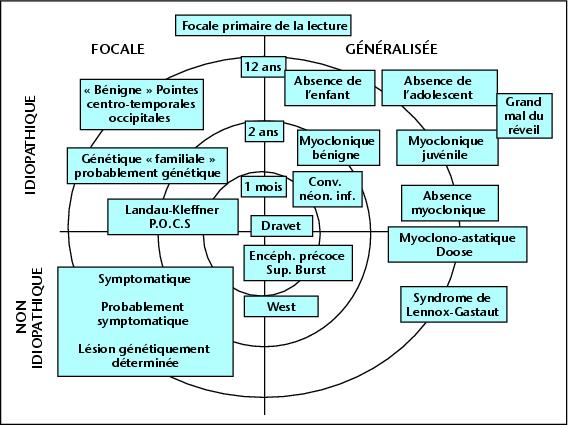
On distingue 3 groupes étiologiques :
– Causes symptomatiques (d’origine structurelle ou métabolique)
– Epilepsie idiopathique, d’origine génétique présumée, avec un pronostic généralement favorable
– Epilepsie cryptogénique : cause indéterminée après investigations
Ils sont classés cliniquement en 2 groupes selon le type de crise : généralisée ou partielle (=focale)
A) Crise généralisée
- Causes symptomatiques
| Etiologie | Clinique | Paraclinique |
|---|---|---|
| Cause toxique et métabolique* | Surtout crise tonico-clonique | Bio |
| Syndrome de West = maladie des spasmes en flexion (symptomatique ou idiopatique) | Révélation entre le 4e et le 7e mois (3/1000 naissances) – Crises : spasmes en salves (flexion > extension) – Régression psychomotrice : indifférence, n’apprend plus, puis perd ses acquisitions antérieures |
EEG (pathognomonique) : hypsarythmie (ondes très amples et lentes, pointes diffuses, irrégulières et permanentes) interrompue lors des spasmes par un aplatissement transitoire (suppression burst) |
| Syndrome de Lennox-Gastaut (symptomatique ou cryptogénique) |
Début avant 8 ans, pic à 3-5 ans, parfois dans les suites d’un syndrome de West – Crises toniques, atoniques, absences atypiques : quotidiennes et coexistant chez un même patient – Retard intellectuel, troubles mentaux, de la personnalité, parfois traits autistiques ou prépsychotiques |
EEG intercritique: nombreuses pointes-ondes lentes (1,5-2 Hz), en bouffées bisynchrones ± symétriques, et décharges de rythmes rapides recrutants au cours du sommeil correspondant à des crises toniques pathognomoniques |
| Cause lésionnelle | (cf ci-dessous) Crise focale mais souvent d’apparence généralisée tonico-clonique |
IRM |
* Causes toxiques
– Alcool : prise inhabituelle d’une grande quantité, sevrage, ou épilepsie alcoolique (= survenue de crises peu fréquentes chez un éthylique chronique)
– Cocaïne
– Amphétamines
– Plomb, manganèse, organophosphorés…
Causes médicamenteuses
– Psychotropes : sans surdosage (imipraminiques, tramadol), par surdosage (lithium, antidépresseurs), par sevrage (BZD, barbituriques)
– Autres (médicaments « convulsivants ») : théophilline, ciclosporine, isoniazide, méfloquine
Causes métaboliques
– Hypoglycémie, hypocalcémie, hyponatrémie : crises généralisées tonicocloniques
– Insuffisance rénale : myoclonies
– Déficits en vitamines B (nouveau-né ++)
(Hyperglycémie avec hyperosmolarité => crises partielles sérielles)
- Epilepsies idiopathiques
| Etiologie | Clinique | Paraclinique |
|---|---|---|
| Epilepsie-absence de l’enfant et de l’adolescent | 10 % des épilepsies entre 3 et 12 ans, pic de fréquence à 7 ans, prédominance féminine Absences typiques, fréquentes (parfois > 100 /j), facilement provoquées par l’hyperpnée Bon pronostic immédiat, mais possible évolution en crises généralisées tonicocloniques isolées ou associées aux absences (40%) Chez l’ado : absences moins fréquentes, crises tonicoclonique associées |
EEG : pointes-ondes bilatérales régulières de début et fin brusques à 3 Hz, interrompant un tracé normal |
| Epilepsie myoclonique juvénile bénigne | Début à l’adolescence Secousses myocloniques en pleine conscience, plutôt matinales (signe de la tasse de café), favorisées par le manque de sommeil, les réveils brusques, la photosensibilité Epilepsie pharmaco-dépendante : 90 % de récidive à l’arrêt du traitement |
EEG intercritique : polypointes-ondes généralisées, souvent avec photosensibilité
(Génétique identifiée : 2 gènes majeurs sur K6 et K15) |
| Epilepsie avec crise généralisée tonicoclonique du réveil | Début à l’adolescence ou chez le jeune adulte, prédominance féminine Crises favorisées par le manque de sommeil, l’absorption excessive d’alcool, le réveil provoqué, la photosensibilité Souvent sensible à une monothérapie |
EEG intercritique: pointes-ondes ou pointes généralisées |
| Syndrome de Dravet (le plus souvent mutation de novo SCN1A) 1B | Début dans le 1ère année de vie par des crises prolongées en contexte fébrile ou post-vaccinal Répétition sous forme d’états de mal peu ou pas fébriles, généralisés ou hémi-corporels jusqu’à 2 ans, puis crises focales, absences atypiques ou myoclonies juvéniles Déficit intellectuel souvent associé Epilepsie pharmaco-résistante |
|
| Syndrome de West ((idiopathique une fois sur 3, meilleur pronostic) |
cf ci-dessus |
B) Crises partielles = focales
- Etiologies symptomatiques
| Etiologie | Clinique | Paraclinique |
|---|---|---|
| Causes lésionnelles* | Clinique souvent évocatrice Crises partielles +/- secondairement généralisées (voir d’apparence généralisée d’emblée) |
IRM ++ |
| Epilepsie de la face interne du lobe temporal avec sclérose ou atrophie hippocampique | Début chez l’adolescent ou le jeune adulte, ATCD de convulsion fébrile compliquée dans l’enfance Crises temporales simples ou complexes groupées avec intervalle libre variable, généralisation secondaire rare.tmm Séquence typique: gêne épigastrique ascendante, état de rêve, arrêt moteur avec automatismes, et attitude dystonique d’un membre controlatéral |
EEG : corrélé à la clinique IRM : atrophie ou sclérose hippocampique |
| Hyperglycémie osmolaire | Glycémie |
* Causes lésionnelles
> Tumorales : 10 à 15 % des épilepsies de l’adulte, rare chez l’enfant. Surtout les tumeurs d’évolution lente impliquant précocement le cortex (astrocytome de bas grade, oligodendrogliome, méningiome)
> Vasculaires : accidents hémorragiques > ischémiques (survenue à la phase aiguë ou tardivement sur cicatrice séquellaire), malformations vasculaires
> Traumatiques
– Crises précoces (< 7j) : réactionnelles, FdR d’épilepsie
– Crises tardives (> 7j, répétition spontanée) : épilepsie post-traumatique (rare, installation en moins de 2 ans la plupart du temps)
> Infectieuses (encéphalites, méningo-encéphalites, abcès cérébraux) : début à tout âge, les causes les plus fréquentes sont l’encéphalite herpétique, et la neurocysticercose en zone tropicale
> Dysplasie corticale focale et autres anomalies du développement cortical (erreurs de migration neuronale) : début à tout âge, épilepsie pharmaco-résistante ± retard mental, signes neuro et ATCD familiaux associés
- Epilepsies partielles idiopathiques (uniquement chez l’enfant et l’adolescent)
| Etiologie | Clinique | Paraclinique |
|---|---|---|
| Epilepsie à paroxysmes rolandiques ou à pointes centro-temporales | Début entre 3 et 13 ans, prédominance masculine Crises partielles simples de la région buccofaciale (clonies, paresthésies, hypersalivation, impossibilité de parler en pleine conscience), très liées au sommeil ± extension au membre supérieur ou généralisation secondaire Guérison vers 16 ans |
EEG intercritique : pointes centro-temporales lentes biphasiques, sur rythme de fond normal |
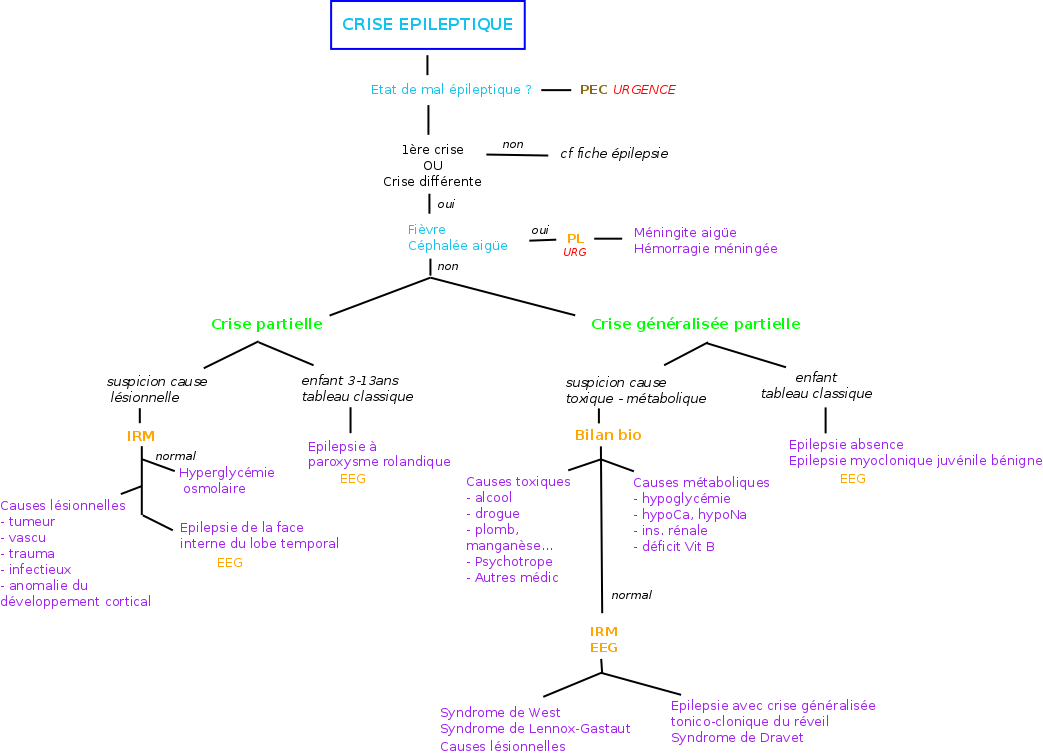
L’OD est à réaliser dans le cas d’une première crise ou lors d’une crise atypique chez un épileptique connu
A) Clinique
Anamnèse : Interrogatoire du malade et des témoins à la recherche
– Des signes paroxystiques, leur séquence chronologique (crise focale/généralisée ?)
– D’autres types de crises
– D’antécédents personnels ou familiaux orientant vers un syndrome épileptique.
Clinique : Recherche de signes d’étiologie lésionnelle ou non : focalisation neurologique, syndrome méningé, affection médicale non-patente
B) Paraclinique
Biologie : si suspicion de cause métabolique ou toxique
– Glycémie
– Ionogramme
– Fonction rénale et hépatique
– Alcoolémie
Imagerie cérébrale (IRM en première intention, TDM en attendant si IRM non dispo. Possible à distance si l’examen neuro est normal) Systématique sauf intégration certaine de la crise dans un de ces 3 syndromes non-lésionnels :
– Epilepsie-absence
– Epilepsie myoclonique juvénile bénigne
– Epilepsie à paroxysmes rolandiques
EEG (cf fiche Epilepsie) : meilleure rentabilité à 24-48h de l’épisode aigu, en l’absence de traitement anti-épileptique
PL : si céphalée aiguë avec scanner normal (épistaxis méningée), ou fièvre (méningo-encéphalite)
C) Synthèse 0
Cf fiche MGS : Crise épileptique




3 réponses à “Crise épileptique”
Les termes des épilepsies idiopathiques « petit-mal » (absence) et « grand-mal » (crise tonico-clonique) ne semblent plus exister
J’ai toujours du mal à comprendre vraiment la différence entre idiopathique (=on n’a pas trouvé de cause) et cryptogénétique (=on n’a pas trouvé de cause… mais peut-être qu’il y en a une)
Je ne trouve aucune trace de l’épilepsie avec crise généralisée tonicoclonique du réveil („grand mal du réveil“) sur le net…