
Onco – Hémato – Rhumato – Néphrologie
Fiche réalisée selon le plan MGS
Item ECNi 317
Déf : le myélome multiple (MM), ou maladie de Kahler, est une hémopathie maligne caractérisée par le développement d’un clone de plasmocytes tumoraux de type B envahissant la moelle hématopoïétique. Cette hémopathie est presque toujours non curable.
Epidémio
– Incidence (Fr) = 4000 cas / an, 1 % de l’ensemble des cancers, 10 % des hémopathies malignes
– Plus fréquent avec l’âge (moyenne de 70 ans au diagnostic), n’existe pas chez l’enfant
– Le syndrome POEMS survient surtout chez l’homme vers 50 ans 1B
| Clinique / Paraclinique |
|---|
| Forme habituelle : association infiltrat plasmocytaire médullaire (myélogramme indispensable) + Ig monoclonale dans le sérum et/ou les urines + atteinte osseuse |
A ) Clinique
- Facteurs de risque
Les causes du MM sont inconnues. Il est toujours précédé d’un état « prémyélomateux » de type gammapathie monoclonale d’apparence bénigne ou indéterminée (MGUS), avec un risque annuel de 1 % d’évolution en MM.
- Présentations habituelles
Découverte fortuite chez un patient asymptomatique (> 20%)
Formes symptomatiques
– AEG
– Douleurs osseuses (> 80%) intéressant le rachis, les côtes, le bassin, plutôt d’allure mécanique 1B
– Tuméfaction osseuse (plasmocytome) possible
– Révélation par une complication : signes de cytopénie (anémie ++), fracture pathologique, insuffisance rénale, hypercalcémie, complications osseuses ou infectieuses…
Notes
– Absence de fièvre en dehors de la phase terminale ; pas de tuméfaction des organes hématopoïétiques habituellement.
– La forme habituelle de myélome multiple symptomatique valide 1 ou plusieurs critères de l’acronyme anglais CRAB (hyperCalcémie, insuff. Rénale, Anémie, Bone disease)
– 3 critères de malignité ont été rajoutés aux critères CRAB pour le diagnostic de MM symptomatique 1B : ≥ 1 lésion focale ≥ 5 mm en IRM ; plasmocytose médullaire ≥ 60% ; ratio chaînes légères libres impliquées / non-impliquées ≥ 100.
- Formes cliniques
Formes selon l’Ig monoclonale
MM à chaînes légères isolées : évolution fréquente en insuffisance rénale
MM à IgD (2%) : chaîne légère presque toujours de type λ, mauvais pronostic avec insuffisance rénale, hypercalcémie et amylose
MM non-excrétants (2%)
Plus rarement
– MM biclonaux
– MM à Ig précipitant à basse température (cryoglobuline)
– Exceptionnellement, MM à IgM ou IgE
Myélome multiple asymptomatique
– Défini par une protéine monoclonale > 30 g/L et/ou un envahissement médullaire > 10 % de plasmocytes, mais sans atteinte d’organe (absence de critère CRAB), aussi appelé indolent ou à faible masse tumorale
– Evolution vers un myélome multiple symptomatique après un délai variable, parfois plusieurs annéesPlasmocytome solitaire
Définition
– Une lésion ostéolytique plasmocytaire avec absence d’infiltration plasmocytaire médullaire en dehors de ce site
– Des radiographies osseuses et une IRM normales en dehors de l’unique lésion lytique
– L’absence ou un taux faible d’Ig monoclonale sérique et/ou urinaire, sans effondrement des autres classes d’Ig.
Evolution vers l’apparition de nouvelles lésions lytiques ou d’un authentique myélome multiple. Il existe aussi des plasmocytomes solitaires extra-osseux (nasopharynx, sinus) qui ont une tendance moindre à la dissémination.
Leucémie à plasmocytes
Présentation proche d’une leucémie aiguë, avec
– Anémie et thrombopénie sévères
– Plasmocytose > 2 G/L ou 20 % des leucocytes
– Hépato-splénomégalie et fièvre
Le pronostic reste médiocre malgré les traitements actuels.
Myélome de forme décalcifiante diffuse (5-10%) 1B : déminéralisation diffuse parfois isolée, justifiant la recherche d’un myélome devant toute ostéoporose d’allure commune.
Myélomes ostéocondensants (3% 1B)
Association à une polyneuropathie (30-50 % contre 3 % des autres myélomes multiples) sensitivo-motrice, diffuse, progressive ; s’intégrant parfois dans le cadre plus général d’un syndrome POEMS (Polyneuropathie, Organomégalie, Endocrinopathie, protéine Monoclonale, lésions cutanées – Skin).
B ) Paraclinique
- Biologie
VS
– Très augmentée (> 100 mm) dans 85 % des cas, parfois peu augmentée ou normale dans les MM à chaînes légères, les MM non-excrétants, et les cryoglobulinémies
– Sa mesure itérative n’a pas de réel intérêtHémogramme
– Normal
– Anémie normochrome normocytaire arégénérative ++ : avec rouleaux érythrocytaires au frottis, mécanismes multiples (infiltration médullaire massive, insuffisance rénale, déficit relatif en EPO, effet des cytokines…)
– Leucopénie et thrombopénie : rares et de mauvais pronostic, reflétant une masse tumorale importante
– Pancytopénie dans les formes évoluées (masse tumorale, chimio)
– Plasmocytes circulants exceptionnels au diagnostic (leucémie à plasmocytes, < 2 %)Electrophorèse des protéines sériques et urinaires (EPS et EPU) avec immunofixation
Protidémie totale souvent élevée (Ig monoclonale)
EPS
– Pic à base étroite dans la région γ, β ou plus rarement α2-globulines (80%), associé à une diminution de la quantité d’Ig en région γ
– Agammaglobulinémie isolée dans le myélome multiple à chaînes légères
– Absence de pic dans le myélome multiple non-sécrétant ou non-excrétant
Note : dans ces 2 dernières situations (MM à chaînes légères et MM non- ou peu excrétants), il est recommandé de doser les chaînes légères libres du sérum.
Dosage pondéral des Ig : ne doit plus être réalisé.
Immunofixation : typage de l’Ig monoclonale pour ses chaînes lourde et légère
– IgG 65 %
– IgA 15 %
– IgD 1 %, IgE exceptionnelle 1B
– Type urinaire pur (à chaînes légères) 15 % : type κ (2/3) ou λ (1/3) indépendamment de l’isotype
– Variants rares 5 %
EPU : protéinurie de Bence-Jones (90%), constituée de chaînes légères κ ou λ
Myélogramme (± BOM si non-contributif) – indispensable au diagnostic
Présence d’un infiltrat plasmocytaire (> 10 % d’éléments nucléés 1A, 1B ; ou < 10% avec importantes dystrophies nucléaires 1B)
Anomalies morphologiques des plasmocytes : peuvent être observées, mais pas indispensables au diagnostic
Analyse cytogénétique des plasmocytes (FISH) : impact pronostique majeur !
- Imagerie
Bilan osseux systématique comprenant des clichés du crâne, rachis, complet, bassin, thorax et grils costaux, humérus et fémurs. A tout moment, une douleur osseuse brutale justifiera la réalisation d’une nouvelle radio sur le site douloureux (géodes ‘à l’emporte-pièce’ 1B).
Techniques d’imagerie
– La radiologie conventionnelle n’est plus la référence !
– IRM : systématique, permet le diagnostic des complications ostéoneurologiques (compression médullaire ou radiculaire…) 1A, diagnostic des plasmocytomes solitaires et de certaines fractures vertébrales 1B
– TDM corps entier sans injection : préférable aux radio standard (indiquée si CI à l’IRM, guidage de biopsie si extension aux parties molles… 1B)
– PET-scan : place d’importance croissance dans le diagnostic 1A, localisation extra-médullaire 1B
– Pas d’indication à la scintigraphie osseuse
Aspect radiologique
– Ostéoporose, lésions ostéolytiques (géodes ou lacunes, prédominant dans les régions de forte hématopoïèse) et fractures, souvent associées
– Selon la localisation : tassement « en galette » du rachis, géodes à l’emporte-pièce (sans liseré de condensation périph.) ± fractures sur les os longs, courts et plats
– Pas de lésion en radio standard (10-20%)
Note : la reminéralisation des lésions osseuses spécifiques sous traitement est rare, y compris chez les patients répondeurs.
C ) Diagnostic différentiel
Ostéoporose sévère, cancer secondaire des os (pas de clone dans le sang ou les urines)
MGUS et myélome multiple à faible masse tumorale : aucun moyen ne permet à ce jour de certifier le caractère bénin d’une dysglobulinémie monoclonale, seule l’évolution permettra de trancher.
Il existe tout de même des éléments d’orientation vers le MGUS
– Taux d’Ig plutôt faible (< 30 g/L)
– Protéinurie de Bence-Jones nulle ou minime
– Plasmocytose médullaire faible (< 10%) faite de plasmocytes non-dystrophiques
– Pas de douleur osseuse, de lésion ostéolytique, d’anémie, d’insuffisance rénale ni d’hypercalcémie
L’évolution 1B d’un MGUS peut se faire vers une hémopathie maligne (myélome multiple, rarement une maladie de Waldenström, un lymphome non-Hodgkinien ou une LLC) avec un risque annuel de 1 %, risque d’autant plus important que l’Ig n’est pas de type G, et que le rapport κ / λ est anormal.
Le suivi 1B hématologique est réalisé à vie (EPS biannuel puis annuel), il est adapté à la présence ou non des FdR de progression au diagnostic, et de l’évolution (stabilité ou augmentation).
Maladie de Waldenström 1B
– Définie par une prolifération lymphoplasmocytaire clonale médullaire + sécrétion d’une dysglobuline monoclonale de type M ± lymphocytose sanguine clonale
– Signes généraux possibles : hypertrophie ganglionnaire, splénique ou hépatique, amaigrissements, sueurs, anémie de mécanismes divers
– ± Syndrome d’hyperviscosité (céphalées, vertiges), signes auto-immuns (cryoglobulinémie, neuropathie avec anti-MAG, hémolyse)
Autres causes plus rares d’Ig monoclonale 1B
– LLC
– Lymphome non-Hodgkinien de type B
– Amylose
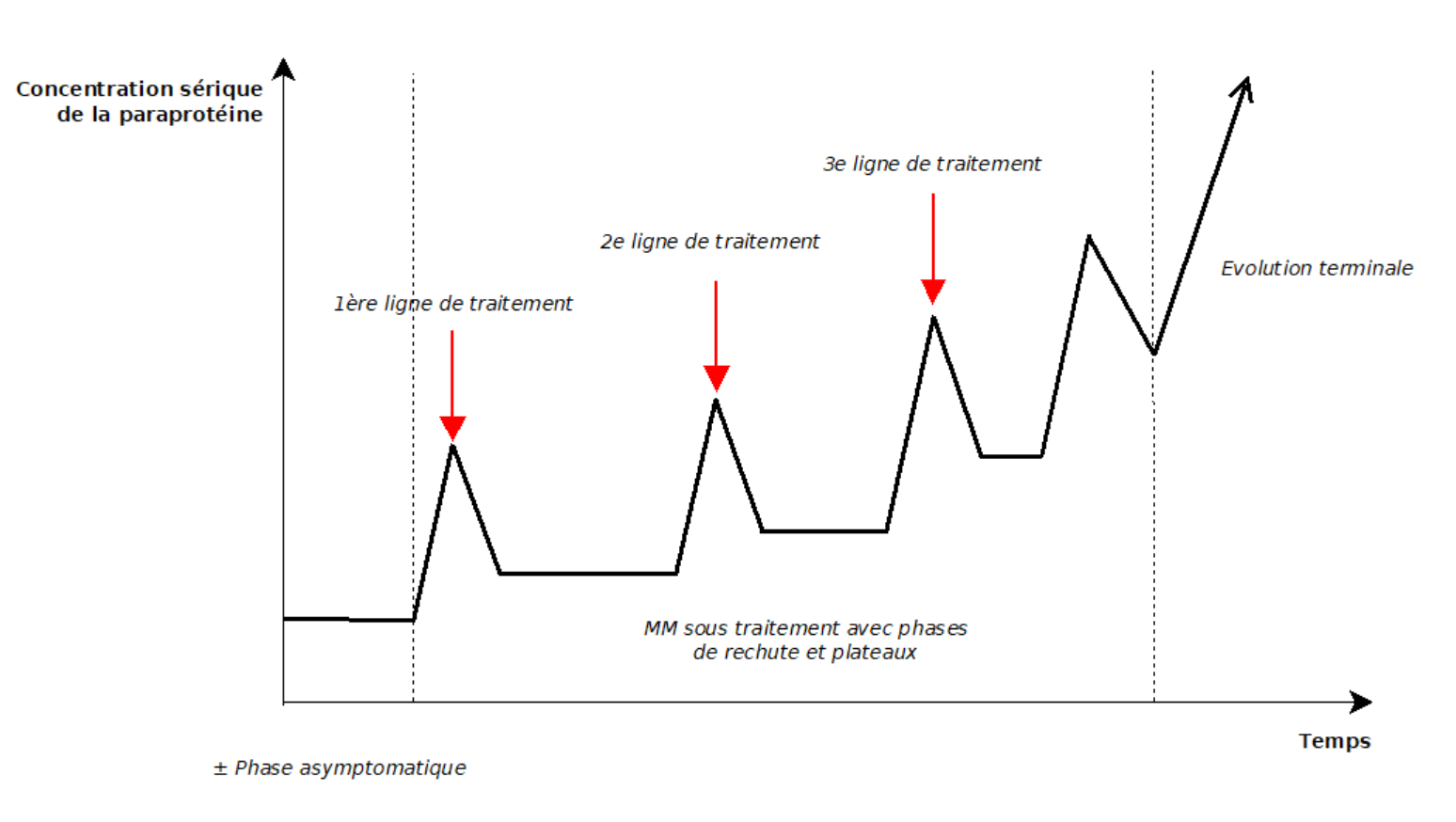
A ) Histoire naturelle
L’évolution sous traitement est une alternance de
– Phase de plateau durant 12 à 18 mois pour la première, correspondant à une diminution de l’activité proliférante de la tumeur et durant laquelle la chimiothérapie est arrêtée (préjudiciable car elle n’améliore pas le pronostic et favorise les SMD).
– Systématiquement suivie d’une rechute, avec reprise de la chimiothérapie
1 à 4 phases de plateau séparent le diagnostic du décès, avec des réponses au traitement de plus en plus rares et courtes, et des complications plus graves et fréquentes.
B ) Complications
Complications osseuses 1B
– Fractures pathologique : très évocatrices au rachis si > T5
– Compressions radiculaires ou médullaires
– Hypercalcémie (10 %) : le plus souvent symptomatique, facteur de mauvais pronostic
Insuffisance rénale (50 % dans l’évolution) 1B : 4 groupes de mécanisme différent
Néphropathie tubulo-interstitielle = rein myélomateux (80 % des IRA au cours du MM)
– Précipitation intra-tubulaire de chaînes légères et de protéine de Tamm-Horsfall conduisant à une rupture de la membrane corticale
– Plus fréquent dans les MM à chaînes légères (sans prédominance d’un isotype particulier), et les MM à IgD 1C
– Précipitation favorisée par la déshydratation (hypercalcémie, traitements, infections), les néphrotoxiques surajoutés (AINS, IEC) et les PdC iodés
– PBR 1C : indiquée en l’absence de facteur déclenchant ou dans les situations pathologiques complexes, montre des cylindres obstruant les tubules distaux et collecteurs (polychromatophiles, aspect fracturé et réaction giganto-cellulaire de contact), altérations de l’épithélium tubaire, fibrose interstitielle ± importante
Syndrome de Fanconi (rare)
– Tubulopathie proximale (ART type 2 1C) par toxicité particulière des chaînes κ sur les cellules tubulaires
– Glycosurie avec glycémie normale, aminoacidurie généralisée, hypophosphatémie (± compliquée d’ostéomalacie 1C), acidose chronique 1B, hypouricémie, insuffisance rénale dans 1 cas / 2 1C
Néphropathie par dépôts d’Ig
– Amylose AL (5-10%) : surtout myélome à chaînes légères λ, localisations tissulaires, rénales, cardiaques, neurologiques, synoviales
– Syndrome de Randall = maladie des chaînes légères : les chaînes légères en excès ne prennent pas de structure fibrillaire, tableau de néphropathie glomérulaire avec protéinurie voire syndrome néphrotique
– PBR 1C : indiquée si albuminurie > 1 g/24h
IRA fonctionnelle par hypercalcémie 1C
Infections récidivantes
– 1ère cause de décès en cas de MM 1A
– En particulier infections ORL et pulmonaires 1B
Insuffisance médullaire 1B
– Anémie de divers mécanismes : hémodilution liée à la gammapathie, insuffisance médullaire quantitative, insuffisance rénale, auto-immunité
– Autres cytopénies et pancytopénie possibles, cf. partie 2B
Amylose (5-15 %) : manifestations neuro, rénales, cardiaques, synoviales (canal carpien)
SMD et leucémies secondaires (2-3%), favorisés par l’usage prolongé d’agents alkylants
Syndrome d’hyperviscosité (rare)
Phénomènes auto-immuns 1B
– Cryoglobulinémie : précipitation au froid
– Anémie hémolytique, autres
C ) Pronostic
Facteurs pronostiques : un âge élevé est le seul facteur lié à l’hôte, les autres facteurs pronostics sont liés à la tumeur
| Groupe | Facteurs pronostiques |
|---|---|
| Masse tumorale | Anémie / thrombopénie Lésions lytiques étendues Créatinine sérique élevée |
| Malignité intrinsèque | Anomalies chromosomiques t(4;14), del(17p) 1A, 1B, del13 1B β2-microglobuline élevée Albumine sérique basse LDH élevée Cytologie plasmoblastique |
| Traitement | Chimiorésistance |
Scores pronostiques
Index pronostique international (2005) : décrit 3 stades de gravité selon la β2m, le LDH, l’albuminémie et le profil moléculaire. Ce test n’est valable qu’au diagnostic, il n’est pas validé lors du suivi et lors des rechutes.
| Stade | Critère | Survie médiane 1B |
|---|---|---|
| I | Tous les marqueurs sont normaux β2m et LDH normaux Absence d’anomalie t(4;14), del(17p) |
62 mois |
| II | Ni I ni III | 45 mois |
| III | β2m ≥ 5,5 mg/L et LDH élevés ou présence d’une anomalie t(4;14), del(17p |
29 mois |
Note 1B : le syndrome POEMS est de meilleur pronostic, avec une moyenne de survie de 165 mois, d’autres formes sont plus graves comme la leucémie avec plasmocytes circulants, rapidement létale.
Classification de Durie et Salmon : totalement abandonnée 1A / toujours présentée dans le référentiel de rhumatologie 2018 1B
Survie : de l’ordre de 8-10 ans pour un diagnostic avant 65 ans, ou de 5-6 ans chez les patients plus âgés.
A ) Bilan
Bilan d’extension (bilan osseux) : cf. partie 2, imagerie
Bilan des complications
– NFS, créatininémie, ionogramme avec calcémie
– Bilan d’hémostase (manifestations hémorragiques par thrombopathie fonctionnelle, exceptionnellement troubles de la coagulation)
Appréciation pronostique : dosages sanguins β2-microglobuline, albumine, LDH (« la CRP n’a pas ou plus d’intérêt dans le MM »)
B ) Traitement
- Traitement anti-tumoral
Myélome multiple symptomatique
Molécules disponibles
– Alkylants : melphalan, cyclophosphamide
– Corticoïdes : dexaméthasone, prednisone
– Immunomodulateurs : thalidomide, lénalidomide, pormalidomide
– Inhibiteurs du protéasome : bortezomib, carfilzomib, ixazomib
– Ac monoclonaux anti-CD38 : daratumumab, isatuximab
– Biphosphonates : pamidronate, acide zolédronique (peuvent être associés au long cours à la chimiothérapie pour réduire l’atteinte osseuse)
Indications
– Jusqu’à 65 ans, traitement intensif par Melphalan forte posologie et auto-greffe de CSH du sang périphérique
– Après 65 ans, le traitement est adapté, ces patients souvent non-éligibles à l’auto-greffe reçoivent généralement une chimiothérapie par MPT (melphalan-prednisone-thalidomide), MPB (melphalan-prednisone-bortezomib) 1A, 1B ou RD (lénalidomide-dexaméthasone) 1B.
Myélome multiple asymptomatique
Une traitement n’est pas indiqué dans cette situation, mais ces patients font l’objet d’une surveillance clinico-biologique attentive, la chimiothérapie devient indiquée en cas d’évolution vers un MM biologiquement actif.
Plasmocytome solitaire (osseux ou extra-osseux)
La radiothérapie est le traitement de choix, venant parfois compléter une exérèse chirurgicale ± complète.
- Traitement symptomatique
Anémie : PEC par EPO recombinante ou transfusions
Infections : ATBthérapie précoce, éviter si possible les néphrotoxiques (± prophylaxie par perfusion d’Ig polyvalente 1B)
Douleurs osseuses : antalgiques de niveau adapté ± radiothérapie locale d’un foyer tumoral douloureux, chirurgie orthopédique préventive d’une lésion lytique à haut risque sur un os long
Epidurites et compressions médullaires (urgences!)
– Bilan : IRM et avis neurochirurgical
– Puis décision d’une PEC chirurgicale (laminectomie) ou par radiothérapie + corticothérapie
Insuffisance rénale
– Prévention : hydratation, CI aux AINS, limiter les médicaments néphrotoxiques et injections de produit de contraste iodés 1A
– Mesures supplémentaires 1B : alcalinisation des urines pour le rien myélomateux, traitement symptomatique d’un sd de Fanconi (correction du pH, phosphate, supplémentation vitD3)
– Recours à la dialyse rare
Hypercalcémie (urgence!) : hydratation, biphosphonates (également indiqués en prophylaxie 1B)
Syndrome d’hyperviscosité : échanges plasmatiques (plasmaphérèse), mise en route rapide du traitement hématologique spéficique
- Suivi
La réponse au traitement est évaluée sur la disparition des signes cliniques, et la réduction du taux de la protéine monoclonale sérique et/ou urinaire. La réponse complète se définit par une normalisation de la plasmocytose médullaire et la disparition du composant monoclonal en immunofixation.
EPS et EPU sont des éléments très importants du suivi thérapeutique, mais il est inutile de multiplier les immunofixations (en dehors du diagnostic initial et en vue d’une fin de traitement).
Le terme de guérison peut être avancé avec prudence chez de rares patients ayant reçu une allo-greffe de CSH. Les traitements intensifs récents utilisés chez des patients n’ayant pas de facteur pronostique défavorable pourraient permettre des survies très prolongées.




3 réponses à “Myélome multiple des os”
Selon le collège d’hématologie 2018:
– Le dosage pondéral des Ig n’est plus recommandé
– La CRP n’est plus nécessaire (même dans les facteurs pronostics)
– La radio conventionnelle n’est plus la référence et l’IRM est systématiquement réalisée (en désaccord avec la rhumato …)
– La classification de Dulrie et Salmon n’existe plus
Le plasmocytome solitaire est-il un type de myélome multiple, ou un dg. diff ?! La définition n’est pas très précise.
Bonsoir , svp au cour du myelome les tassements vertebraux sont considéré comme des fractures pathologiques ? Et comptabilisé comme événements osseux ou non .merci pour aide